
Hauptproblem: Abnehmende Lungenfunktion
Cystische Fibrose (Mukoviszidose) kann sich auf alle Organe auswirken, die für ihre Funktionsfähigkeit auf CFTR*-Proteine angewiesen sind.1,2,3 Menschen mit CF können von einer großen Bandbreite an Symptomen betroffen sein.4 Wer wie stark an welchem Symptom leidet, hängt von den verursachenden CFTR-Mutationen des bzw. der Einzelnen, der sonstigen genetischen Veranlagung und den äußeren Einflüssen der Umwelt ab.4
Das Leitsymptom der cystischen Fibrose ist die voranschreitende Verschlechterung der Lungenfunktion.5




Atemwege
In der Lunge befindet sich ein Netzwerk aus Bronchien. Das sind feine Röhrchen, die sich immer weiter verästeln, je tiefer man in die Lunge eindringt. Dieses Netzwerk bildet die Atemwege. Sie sind innen von Flimmerhärchen (= Zilien) bedeckt, die sich hin und her bewegen, um Schleim aus den Atemwegen zu transportieren.6
Atemwege bei Menschen ohne CF
Bei gesunden Menschen schützt dünner, wässriger Schleim die Lunge, indem er Krankheitserreger, die Infektionen verursachen können, umschließt. Anschließend wird der Schleim mittels der Flimmerhärchen aus der Lunge befördert, wodurch die Krankheitserreger ausgeschieden werden. Dabei ist es Aufgabe der CFTR-Proteine, dass genug Salz und Wasser in den Schleim gelangt, damit er dünn und wässrig bleibt.6 So regulieren sie den Salz-Wasser-Haushalt.
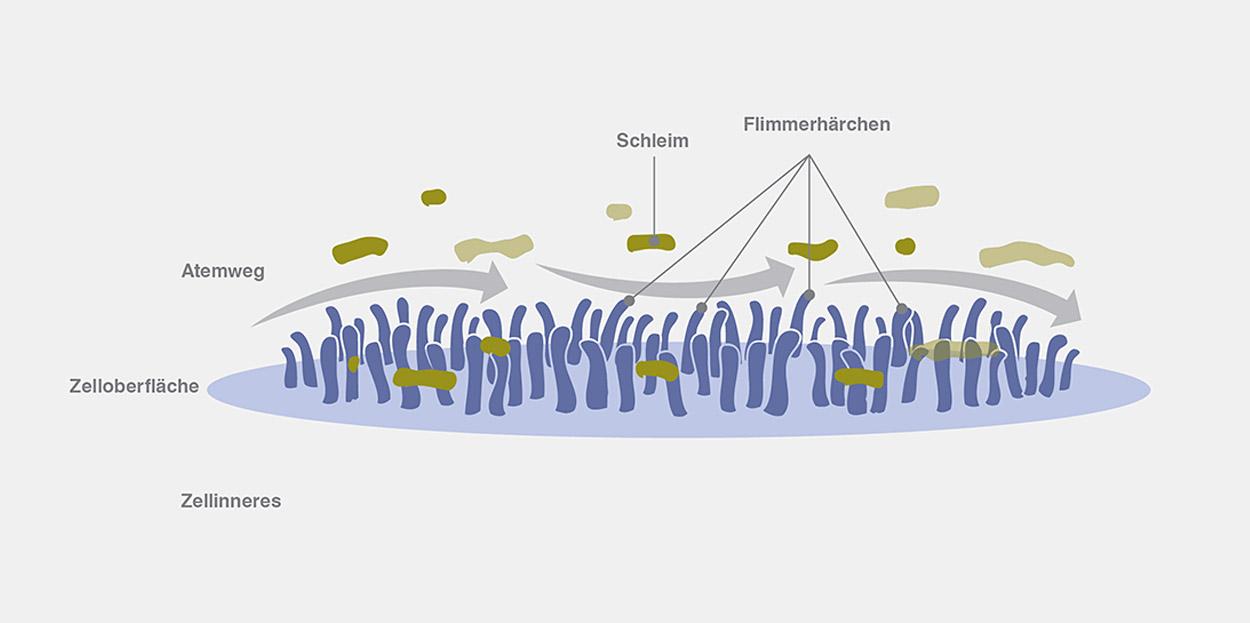
Bei gesunden Menschen können sich die Flimmerhärchen (= Zilien) frei bewegen, sodass der Schleim aus der Lunge abtransportiert werden kann.
Atemwege bei Menschen mit CF
Bei CF-Betroffenen werden deutlich weniger oder nicht funktionsfähige CFTR-Proteine produziert, wodurch der Salz-Wasser-Haushalt gestört wird. Infolgedessen bildet sich in der Lunge anstatt dünnen und wässrigen Schleims dicker und klebriger Schleim.7 Dieser behindert die Bewegung der Flimmerhärchen, sodass diese den Schleim nicht aus der Lunge abtransportieren können. Der Schleim sammelt sich an und verstopft die Atemwege, weshalb dort ein Nährboden entsteht, der das Wachstum von Bakterien begünstigt.8

Bei Menschen mit CF können sich die Flimmerhärchen (= Zilien) aufgrund des dicken, klebrigen Schleims nicht frei bewegen, sodass Schleim und Krankheitserreger nicht aus der Lunge abtransportiert werden können.
Vergleich der Atemwege


Atemwegssymptome
Folgende CF-typische Atemwegssymptome können auftreten:
- Keuchen9
- Kurzatmigkeit9
- Hartnäckiger Husten mit dickem Schleim9
- Lungeninfektionen (durch Bakterien wie Pseudomonas aeruginosa, Burkholderia cepacia und Staphylococcus aureus)5
- Bronchiektasen3 (Schädigung der Atemwege, bei der diese ausgeweitet werden, was mit starkem Husten und großen Mengen schleimigen Auswurfs einhergeht)
Der zähflüssige Schleim kann einen Kreislauf aus Infektionen und Entzündungen verursachen, der über Strukturveränderungen zu einer noch stärkeren Blockade der Atemwege durch ebendiesen Schleim führt. Dieser Kreislauf kann eine nachhaltig veränderte Lungenstruktur bei Menschen mit CF zur Folge haben.3, 10
Tritt eine pulmonale Exazerbation auf, erfordert diese meist eine zusätzliche Behandlung mit Antibiotika (oral, intravenös oder inhalativ). Auch Atemwegstherapien werden in diesen akuten Phasen vermehrt angewandt. Je nach Schweregrad kann auch eine stationäre Aufnahme ins Krankenhaus (Hospitalisierung) erforderlich werden.3

Pulmonale Exazerbationen
Darüber hinaus können die häufigen Infektionen und Entzündungen der Lunge zu pulmonalen Exazerbationen führen.1, 3 Pulmonale Exazerbationen sind Phasen, in denen sich die Symptome der Mukoviszidose in der Lunge deutlich verschlechtern.1, 3 In diesem Zeitraum kann es zu einer verminderten Lungenfunktion, Fieber, vermehrter Schleimproduktion und damit verbundenem (blutigen) Husten sowie Kurzatmigkeit und Gewichtsverlust kommen.1, 3 Menschen mit cystischer Fibrose müssen in diesen Phasen sehr häufig mit Antibiotika behandelt werden, teilweise ist auch ein Krankenhausaufenthalt notwendig.1, 3
Nach der Exazerbation klingen die Symptome komplett oder nur teilweise wieder ab – abhängig davon, ob neue Lungenschäden verursacht wurden.1, 3 Das Risiko für pulmonale Exazerbationen nimmt mit steigendem Alter zu.11 Da sie maßgeblich für eine langfristige Verschlechterung der Lungenfunktion sorgen, zielen viele therapeutische Maßnahmen darauf ab, pulmonale Exazerbationen zu vermeiden und die Lunge möglichst schleimfrei zu halten. Dies erfolgt beispielsweise durch Inhalation von Kochsalzlösungen, schleimlösenden Mitteln oder Antibiotika sowie Atemphysiotherapie.12
Abnahme der Lungenfunktion
Erste strukturelle und funktionelle Veränderungen der Lunge können bereits in den ersten Lebensmonaten nachweisbar sein und sich mit zunehmendem Lebensalter verstärken.13
Frühe strukturelle Lungenveränderungen lassen sich anhand des LCI (Lung Clearance Index) oder mittels bildgebender Verfahren nachweisen, noch bevor eine Beeinträchtigung der Lungenfunktion (Verringerung des Atemvolumens ppFEV1) festzustellen ist.13, 14, 15Diese Lungenschäden können auch dann voranschreiten, wenn die Lungenfunktion noch normal ist.
Der zeitliche Verlauf der Lungenschäden kann sich bei Menschen mit CF stark unterscheiden. In der Regel beginnt der Progress aber während des ersten Lebensjahrzehnts.16

Untersuchungen der Lungenfunktion und -struktur1, 10, 17
Zur Testung der Lungenfunktion gibt es verschiedene Messmethoden:
Spirometrie
Bei der Spirometrie, die auch als „kleiner Lungenfunktionstest“ bezeichnet wird, wird bei verschlossener Nase über ein Mundstück in das Spirometer geatmet. Dieses misst die durchströmende Luftmenge und -geschwindigkeit, sodass das Lungen- und Atemvolumen bestimmt werden kann. Dazu zählt unter anderem der FEV1-Wert, der auch als Einsekundenkapazität bezeichnet wird. Er beschreibt das Atemvolumen, welches innerhalb der ersten Sekunde forciert ausgeatmet werden kann. Die Spirometrie dauert insgesamt nur wenige Minuten.
Ganzkörper-Plethysmographie
Die Ganzkörper-Plethysmographie wird als „großer Lungenfunktionstest“ bezeichnet. Dabei wird in einer Kammer ebenfalls in ein Mundstück ein- und ausgeatmet. Über Druckveränderungen in der luftdichten Kammer können zusätzliche Angaben zum Restvolumen der Lunge sowie zum Atemwegswiderstand getroffen werden. Der große Lungenfunktionstest erfordert eine bessere technische Ausstattung und ist etwas aufwendiger als die Spirometrie.
Strukturveränderungen der Lunge entwickeln sich häufig, bevor eine Verringerung der Lungenfunktion mit klassischen Verfahren messbar ist. Daher können zusätzlich bildgebende Methoden wie Röntgen, Computertomographie (CT) oder Magnetresonanztomographie (MRT) angewandt werden.
*CFTR = Cystic Fibrosis Transmembrane Conductance Regulator
Atemwege bei Menschen mit CF:
Es werden weniger oder nicht funktionsfähige CFTR-Proteine produziert, wodurch der Salz-Wasser-Haushalt gestört wird. Also Folge bildet sich in der Lunge dicker und klebriger Schleim.7

CF-typische Atemwegssymptome:
Keuchen, Kurzatmigkeit, hartnäckiger Husten mit dickem Schleim, Lungeninfektionen und Bronchiektasen.9, 5,3
Pulmonale Exazerbationen:
So nennt man Phasen, in denen sich die Atemwegssymptome verschlechtern, was zu bakteriellen Infektionen, einer reduzierten Lungenfunktion sowie mehr Husten und Schleim und/oder Gewichtsabnahme führt.1, 3
Abnehmende Lungenfunktion:
Bereits in den ersten Lebensmonaten zeigt sich bei CF-Betroffenen eine strukturelle und funktionelle Veränderung der Lunge. Eine solche lässt sich mittels des LCI (Lung Clearance Index) oder bildgebender Verfahren nachweisen.13, 14,15



-
Orenstein D. M.: „Cystic Fibrosis: A Guide for Patient and Family“. 4th ed. Philadelphia, PA: Lippincott Williams & Williams, 2011.
-
Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med 2019; published Online September 27, 2019. https://doi.org/10.1016/S2213-2600(19)30337-6
-
O'Sullivan B, Freedman S, „Cystic fibrosis“, The Lancet, 373, 9768, 1891-1904, 2009.
-
Welsh MJ et al., „Cystic fibrosis: membrane transport disorders“, in Valle D, Beaudet A, Vogelstein B et al. eds. The Online Metabolic & Molecular Bases of Inherited Disease, The McGraw-Hill Companies Inc, www.ommbid.com, 2004, part 21, chap 201.
-
Rowe S et al., „Mechanisms of Disease: Cystic Fibrosis,“ The New England Journal of Medicine, Bd. 352, pp. 1992-2001, 2005.
-
National Heart, Lung and Blood Institute, „The Respiratory System“, [Online]. Available: http://www.nhlbi.nih.gov/health/health-topics/topics/hlw/system. [Zugriff am 06 Oktober 2016].
-
Derichs N, „Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis“, European Respiratory Review, 22, 127, 58-65, 2013.
-
National Heart Lung and Blood Institute, „What is Cystic Fibrosis?“, [Online]. Verfügbar unter: http://www.nhlbi.nih.gov/health/health-topics/topics/cf. [Zugriff am 12 Oktober 2016].
-
Cystic Fibrosis Foundation, „About Cystic Fibrosis“, [Online]. Verfügbar unter: https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/. [Zugriff am 20 Oktober 2016].
-
Naehrig S et al. Cystic fibrosis–diagnosis and treatment. Dtsch Arztebl Int 2017; 114:564–574
-
de Boer K et al., „Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis“, Thorax, 680-685, 2011.
-
Smyth AR et al., „European Cystic Fibrosis Society Standards of Care: Best Practice guidelines“, Journal Cystic Fibrosis, 13 Suppl 1, 23-42, 2014.
-
Sly P. D. et al.: „Lung Disease at Diagnosis in Infants with Cystic Fibrosis Detected by Newborn Screening“. Am J Respir Crit Care Med. 2009;180(2):146–152.
-
de Jong P. A. et al.: „Pulmonary Disease Assessment in Cystic Fibrosis: Comparison of CT Scoring Systems and Value of Bronchial and Arterial Dimension Measurements“. Radiology. 2004;231(2):434–439.
-
Ellemunter H. et al.: „Sensitivity of Lung Clearance Index and Chest Computed Tomography in Early CF Lung Disease“. Respir Med. 2010;104(12):1834–1842.
-
Ramsey B. W.: ProcAm Thorac Soc. 2007;4:359–363.
-
Criée C.-P. et al.: „Leitlinie zur Spirometrie“. Pneumologie. 2015;69:147–164.1